

An Alternative and Sustainable BET Designed for Patient Safety

Animals are essential to enhancing understanding of disease progression and biological mechanisms, as well as drug safety and efficacy, and the components of the 3Rs principle (replacement, reduction and refinement) provide a framework for ethical animal research (1).
A prominent area where sustainability and innovation intersect is the bacterial endotoxin test (BET). Advancements in technology and scientific research have unlocked new possibilities for creating alternatives to animal-derived products. Alongside the established 3Rs framework, an additional consideration for responsible use can also be applied within the pharmaceutical industry, building an additional focus on industry progression by lowering the impact and reliance on animals where possible, ensuring a lens of responsibility for the advancement of science and technology, support for global regulatory bodies and driving innovation for patients.
United States Pharmacopeia (USP) replaced the Rabbit Pyrogen Test (RPT) in 1983 with the Limulus Amebocyte Lysate (LAL) test, sourced from the blood of the Atlantic horseshoe crab, which proved to be more sensitive than RPT (2). This article presents steps to developing a robust, animal-free recombinant cascade reagent (rCR) to replace natural LAL for the BET with a focus on responsibility for patient safety. This kinetic chromogenic assay is designed to simulate the natural LAL reaction. The formulation and composition of rCR assay includes recombinant Factor C, recombinant Factor B and recombinant proclotting enzyme at specific concentrations. The evolution and recombinant cascade provide testing flexibility, efficiencies, confidence and safety in manufacturing injectable and implantable drug products.
rCR Study Stage-Gate Road Map and Decision Process
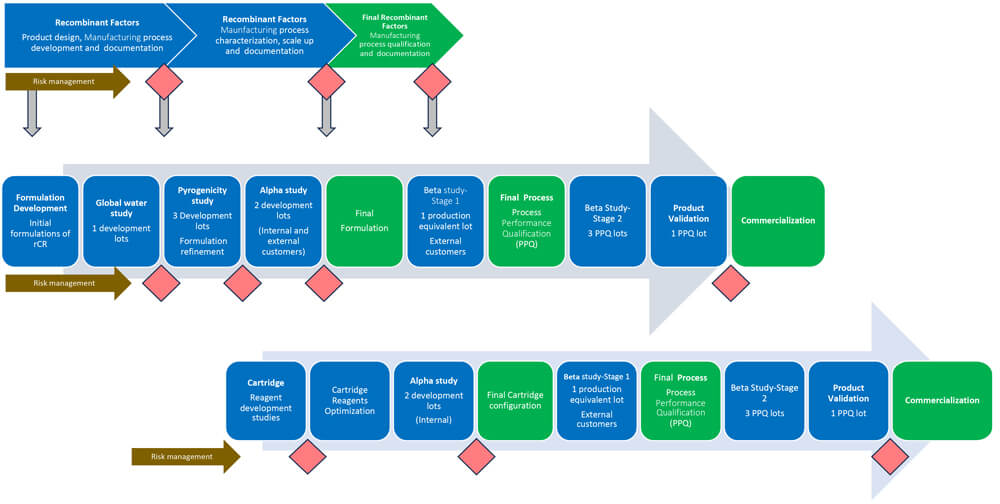
A Stage-Gate methodology for the product development of rCR was applied for scientific rigor and high product quality and to enable data-based decisions. The facilities, manufacturing processes and quality management systems are well established as the product developers were under the Biological License Application for LAL tests. Figure 1 depicts the study roadmap and decision process within the Stage-Gate method, which shows the following below:
- Row 1 – Development of all recombinant factors
- Row 2 – Development of rCR Vials
- Row 3 – Development of Microfluidics Cartridges
Eight Key Considerations for Recombinant Cascade Reagent Development and Study Roadmap
- Risk Analysis and Change Management
The first step was creating and continuously monitoring product risk analysis based on the Failure Modes & Effects Analysis (FMEA) process that allowed thorough identification, prioritization and mitigation or avoidance of the identified risks before these risks are realized by the end-users, and thereby impacting patient safety. The FMEA enhanced the product and process development of rCR vials and microfluidics cartridges by having proper requirements, robust acceptance criteria for tests and product specifications for verification and validation. The product developers have been producing LAL products since 1988, using well-established manufacturing processes under license #1197, approved by the U.S. FDA.
The facility operates under current Good Manufacturing Practices (GMP) as per Code of Regulations Title 21, specifically 210, 211, 600, 601, 610 and 820. Moreover, the change management process was utilized to ensure modifications to existing processes to differentiate LAL and recombinant products were controlled. The FMEA facilitated design alternatives for recombinant factors and identified mitigations linked to stability, manufacturing, facility, in-process testing, quality control release, packaging, and hardware and software for rCR products.
- Recombinant Factors, Scalability and Stability of Cell Lines
As recombinant proteins are a crucial raw material change, it was key that the stability of recombinant factor clones in the chosen cell lines was established to maintain consistency and reproducibility of expression and to reduce the risk of declining protein production and/or enzymatic activity. Cell lines were tested over 60 generations and characterized for sterility, gene copy number, sequence confirmation and stability. Master cell banks were established under robust GMP procedures to ensure a consistent, reliable, high-quality biorepository. The recombinant proteins were manufactured using sterile, endotoxin-free bioreactors and purified in an ISO 13485 and ISO 9001-certified facility. During the scale-up of batches, every batch was tested to ensure compliance with strict specifications and acceptance criteria.
- Variability of Purified Recombinant Factors
Preventing contamination of raw materials and accessories is an essential element of manufacturing process control. To control the variability of purified recombinant factors throughout the manufacturing process, all materials used during cloning, expression, purification and scale-up were endotoxin-free. The quality and consistency of all incoming materials and accessories from external suppliers used in manufacturing were controlled and monitored with appropriate specifications.
- Criticality of Understanding of Patient Safety Factor
To avoid patient level incidents related to endotoxins (3), it is critical to control and test endotoxin levels in the manufacturing processes by recognizing the diversity of endotoxin structures generated by Gram-negative microbes that have adapted to a broad range of parenteral manufacturing conditions. These natural environmental endotoxins (NEEs) exhibit a wide range of biological activity with respect to the LAL test and pyrogenicity and represent parenteral manufacturing contamination risks. As the LAL test is a safety test, any alternative to the LAL test must evaluate the potential safety factor to mitigate patient risks. To calculate a safety factor, a direct comparison of endotoxin detection to the traditional USP <151> Pyrogen Test RPT was conducted using NEEs. Three recombinant formulations, and an FDA-licensed LAL reagent were compared with eight-rabbit RPT fever responses. The estimated calculated safety factor ranges for rCR and LAL were comparable.
- Robustness of Recombinant Formulation
To minimize the risk of underpredicting endotoxins (4), the recombinant formulations required refinement and understanding of the interplay of different recombinant proteins with various factors, including pH, salt ion concentrations, and detergents as these could be present in test articles from customers or manufacturing excipients and impact the activity of the assay (5). The recombinant formulations may provide equivalent results when tested with reference standard endotoxin, but the reactivity could differ from NEEs. According to Figure 1, out of three recombinant formulations, two were selected (formulations 1 and 2) to initiate an alpha study. The objective was to compare the results of endotoxin values obtained with kinetic LAL reagents to rCR vial lots when tested with NEEs in customer samples. The results of inhibition/enhancement in the presence of the product were also compared between LAL reagents and the two rCR vial formulations. Formulation 2 was selected as the final formulation based on the comparable results of NEEs with LAL in water and monoclonal antibody (mAB) samples.
- Collection of Real-World Evidence
Establishing the intended use of a safety test without challenging the design with relevant samples is impossible. Although rCR formulation was refined using an internal collection of samples with NEEs, there was not sufficient data to demonstrate the effectiveness of rCR in detecting endotoxin in a variety of globally sourced in-process or finished products. There could be differences in the microbiota in the environment, samples or water systems based on geographic locations (6). Therefore, a significant effort was made to evaluate endotoxin-positive and endotoxin-negative samples under a global beta study.
For the first stage of the study, a single vial lot was manufactured and released for beta testing. Data was collected per an approved quality assurance protocol with pre‑defined acceptance criteria. Vials were provided to participating customers to compare the inhibition/enhancement profiles between FDA-licensed kinetic LAL reagents and optimized rCR vial products. A direct comparative measurement of NEEs present in the samples was also performed. Samples undergoing routine LAL testing served as test articles. Similarly, one beta rCR microfluidics cartridge lot was shipped to customers to start the first stage with a similar goal to compare rCR cartridges to FDA-licensed LAL microfluidics cartridges using appropriate instruments. Parenteral products from large and/or small intravenous parenteral solutions, small therapeutic molecules, antibiotics, large biological molecules (mAB and plasma products), medical devices and vaccines were targeted.
Three process performance qualification (PPQ) lots for rCR vials and cartridges were produced to qualify the manufacturing process and tested based on pre-approved quality assurance protocols. These lots were used in the second stage of the beta study. LAL results for a specific sample dilution were compared to rCR results at the same concentration. A beta study for rCR vials was conducted first, followed by rCR cartridges, and the number of samples and customers who participated in the studies varied between the two product lines. PPQ lots were also used to verify finished product stability per an established Long Term Stability protocol.
A total of 185 and 228 samples were collected globally for rCR vials and rCR cartridges, respectively. The beta study results demonstrated that for endotoxin-positive samples, both rCR vials and cartridges were equivalent to FDA-licensed LAL products through analysis of the 95% confidence interval for the ratio of rCR to FDA-licensed LAL product endotoxin values. For samples free of detectable endotoxin, the majority obtained acceptable spike recovery at the same dilution as LAL. The data collected indicated that not only are rCR cartridges equivalent to LAL products in endotoxin recovery, but they also exhibited similar interference patterns.
- Product Validation
One PPQ lot for both rCR vials and cartridges was used for product validation. The validation was conducted in accordance with USP methods and procedures described in USP Chapter <85> Bacterial Endotoxin Test, USP Chapter <1223> Validation of Alternative Microbiological Methods and USP Chapter <1225> Validation of Compendial Procedures (7, 8, 9). A full alternate method product validation, including the accuracy, precision, ruggedness and robustness, demonstrated the equivalency of rCR products (vials and cartridges) to FDA-licensed LAL products (vials and cartridges). This is also the case with traditional LAL products, validating the specificity of recombinant testing modalities remains a primary responsibility of the end-user.
- Variability of Recombinant Factors and rCR Products
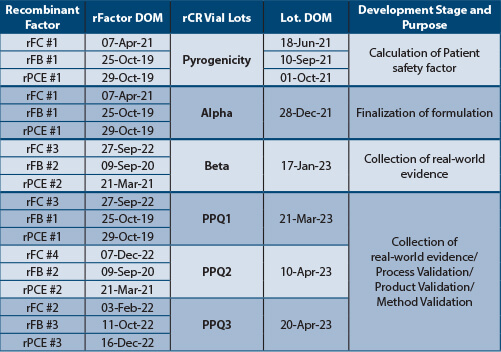
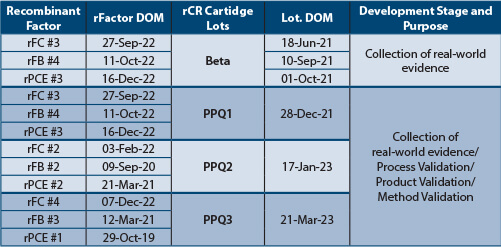
Tables 1 and Table 2 depict the recombinant factors' date of manufacture (DOM) and rCR product DOM to capture the stability of recombinant factors, manufacturing variability of recombinant factors and finished products. The recombinant factors, rCR vials and cartridges lots were manufactured on different days. Each PPQ lot was manufactured using three unique (nine total) lots of recombinant factors. All rCR vial and cartridge lots tested met the acceptance criteria, indicating that recombinant factor lots were stable over time and that the manufacturing process variability was well controlled.
Conclusion
Using a rigorous Stage-Gate process and collecting real-world evidence further highlights the dedication to high-quality standards and continuous improvement in product development. Furthermore, adopting rCR products in the pharmaceutical industry represents a significant step forward in sustainable and ethical testing practices. By reducing reliance on animal-derived products and demonstrating equivalent efficacy and safety to traditional LAL tests, rCR paves the way for more responsible and innovative approaches in medical and pharmaceutical research. This no only aligns with global regulatory trends and the 3Rs principles but also sets a precedent for future advancements in the field, ensuring continued commitment to patient safety and scientific integrity. The ongoing collection of real-world evidence and the refinement of recombinant formulations underscore the importance of adaptive and responsive methodologies in meeting the evolving challenges of endotoxin testing.
References
- Russell, W.M.S., Burch, R.L., The Principles of Humane Experimental Technique, Methuen, London. 1959. Principles - The Johns Hopkins Center for Alternatives to Animal Testing (jhsph.edu)
- USP <151> Pyrogen Test
- 20221021-NatPSA-2022-008-MHRA_Final.pdf (publishing.service.gov.uk)
- Sandle T., Characterizing the Microbiota of a Pharmaceutical Water System-A Metadata Study. 2015 SOJ Microbiol Infect Dis 3(2): 1-8. DOI: http://dx.doi.org/10.15226/sojmid/3/2/00133
- Dubczak, J., Reid, N., Tsuchiya, M., Evaluation of limulus amebocyte lysate and recombinant endotoxin alternative assays for an assessment of endotoxin detection specificity. 2021 Eur. J. Pharm. Sci., 159, 105716. Doi: 10.1016/j.ejps.2021.105716.
- Tsuchiya M., Funatsuki K. Advancement in understanding the criticality of formulation design for recombinant cascade reagent for Bacterial Endotoxins Test for patient safety 2024, manuscript in preparation.
- USP <85> Bacterial Endotoxins
- USP <1223> Validation of Alternative Microbiological Methods
- USP <1225> Validation of Compendial Methods
About the Author
-

Parampal Deol, Charles River Laboratories
Deol is Charles River Laboratories' Vice President of Research and Development for the Manufacturing business division. Deol currently leads the research and development teams, which include system development, system integration, biosciences, technical operations and project management. Deol has more than 30 years of research experience, including 18 years in product development - from feasibility studies to launch and life cycle management using design control and GMP within the medical device and in vitro diagnostic industries. Deol's specialties are risk analysis and managing the safety and efficacy of products and associated manufacturing processes. Moreover, Deol received a post-graduate degree in drug delivery from the Institute of Medical Education and Research in Chandigarh, India, and a master's degree in innovation and marketing management from Poole College of Management at North Carolina State University.