

Isolator Surfaces and Contamination Risks to Personnel GMP Cleaning Requirements for Nonproduct Contact Surfaces

When it comes to protection of cleanroom personnel and product, the possibility for contamination both within and outside an isolator exists.
The issue is of particular interest in the manufacturing of pharmaceutical products with highly potent APIs (HPAPIs). Manufacturers must assess isolator design, the routes by which HPAPI can spread (transfer or contact via nonproduct contact surfaces) and the possibilities for containment with a view to evaluating possible contamination risks within an isolator. Additionally, the cleaning process and cleaning limits for nonproduct contact surfaces within an isolator operated under aseptic conditions, as well as cleaning and air concentration limits outside the isolator, should also be considered.
Validation of the cleanliness of nonproduct contact surfaces has increased in popularity since EMA proposed the following measures to demonstrate effective management of the cross-contamination risk in Chapter 5.21 of Part 1 of the GMP guidelines: “Depending on the contamination risk, verification of cleaning of non product contact surfaces and monitoring of air within the manufacturing area...in order to demonstrate effectiveness of control measures against airborne contamination or contamination by mechanical transfer;...”
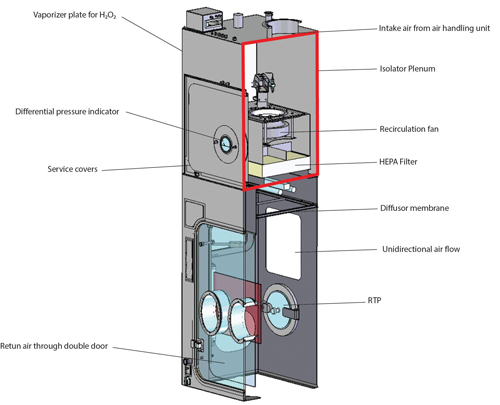
In aseptic manufacturing, isolators are used to reduce direct access by personnel to the critical stages of manufacture (fill–finish) and to contain the cleanroom area in which critical stages take place. As an example, the fill–finish area within an isolator is designed as Zone A/ISO Class 5, and the area outside is Zone D/ISO Class 8 for the European Union and ISO Class 7 for the United States. Protection of personnel handling HPAPIs is another reason for using isolators.
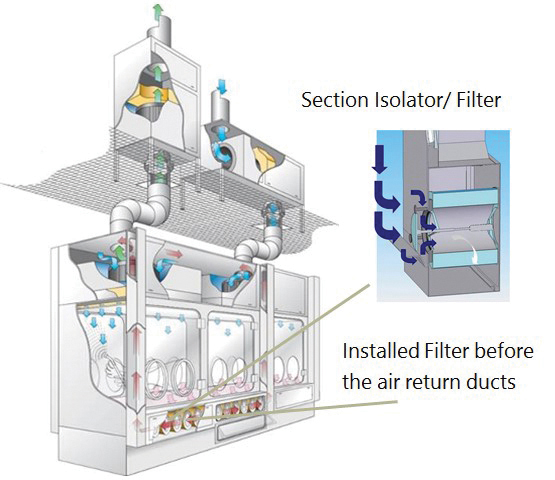
But how does a classic aseptic isolator differ from an isolator used for the aseptic manufacture of HPAPIs? A classic isolator is supplied with conditioned air for Zone A via unidirectional air flow. The return air from the isolator to the recirculation fan travels through the double wall of the isolator (1). Spread of released HPAPIs from the isolator to the isolator plenum is possible; therefore, the classic isolator is not suitable for use of HPAPIs (Figure 1).
An HPAPI isolator, on the other hand, features an additional filter level before the air return into the isolator plenum. This filter level is located directly before the air return ducts, preventing HPAPIs from spreading into return air ducts and the isolator plenum. See Figure 2 for a single-wall isolator with appropriate filter technology between the isolator process chamber and the air return ducts for use with HPAPIs (2).
The primary concern in the manufacture of HPAPIs is to prevent the substance from spreading outside Zone A into the air return ducts and isolator plenum. At the same time, it is also advisable to limit the spread of substances within the isolator chamber; technical measures are required to keep this spread to an absolute minimum. These can include various pressure cascades from the critical manufacturing area to noncritical areas.
The critical HPAPI exposure stages of aseptic manufacturing are the filling process, the fitting of the stoppers and the loading and unloading of the freeze dryer (Figure 3).
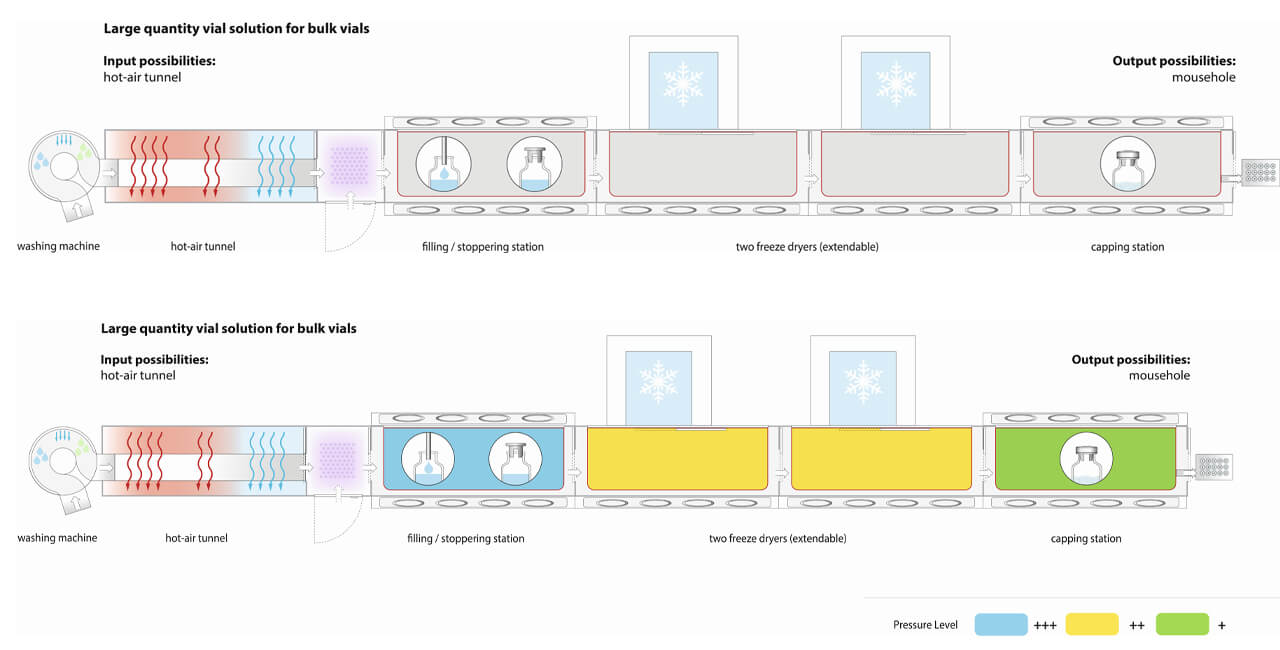
There are various scenarios in which highly potent/toxic substance can spread within the isolator. The key issue here is contamination of the line when a product is being manufactured at that moment. Here, there is no direct risk of cross-contamination, especially as only one product can be manufactured in a given space at any point in time. But if this contamination is not removed effectively during cleaning, this can result in potential cross-contamination risk for the next product manufactured using the same facility.
Product spread can be caused by the breakage of containers such as vials, ampoules, syringes, etc. For the sake of simplicity, this article refers to vials, but this should be understood to include other forms of primary packaging.
The following may cause product spread in an isolator:
- Turbulence due to air flow (pressure cascades) within the isolator line
- Contaminated gloves
- Mechanical transfer systems such as conveyors, carousels, transport carriages for moving equipment to other sections of the isolator, etc.
- Contact contamination due, for example, to damaged vials and gloves, or contaminated stainless steel or plastic surfaces
- Transfer of contaminated settle plates (viable sampler)
So how can these be prevented? The following are some considerations for preventing cross-contamination:
Air Flow
The spread of airborne particles or aerosols can be determined in advance during the planning stage through simulations. These help when it comes to positioning the filters before the air return ducts, and in designing the air flow to the filters. In addition, modifications can be made to the design of areas in which air flow is found to be turbulent.
Mechanical Transfer
During the aseptic fill–finish process, vials, syringes, etc., are transferred using conveyors, separating systems, lifting and transfer systems. These transfer systems can also result in the carryover of highly active substances into neighboring areas. This carryover is critical in the following situations:
- Open filling of vials, syringes, etc. The filling process leads to the release of aerosols that can build up on, dry out and then be released from surfaces/transfer systems/filling equipment such as filling needles
- Breakage of vials. Vial breakage can occur at any time during the manufacturing process and result in contamination of mechanical transfer systems; particularly critical points include separation of the vials, transfer of the vials via carousels, loading and unloading of the freeze dryer, and finally, crimping of the vials
Contact Contamination
Contact contamination within the isolator can be the result of various causes and distribution routes. One common cause is breakage of one or more vials. The following forms of breakage are possible:
- Breakage of a vial contaminates other directly adjacent vials which are then moved on, contaminating more vials on contact
- Breakage of a vial contaminates surfaces such as the floor of the isolator and fixtures within the isolator; if liquids are not removed in a timely manner, they dry up, and the dried active substance may then be spread by the air flow
- Manual cleaning of vial breakage by the operator using gloves attached to the isolator; this can result in contamination of the gloves and cleaning equipment and, depending on where the vial breakage occurs, the route to the next port through which the contaminated cleaning equipment is removed from the isolator can be contaminated
While the forms of contamination mentioned above generally do not yet represent a direct risk of cross-contamination, they can be a challenge in terms of cleaning and should be kept to an absolute minimum. The cleanability of facilities should also be optimized by means of “hygienic design” and by using specific equipment/material which will be changed at the end of a campaign or, at least, prior to a product change to efficiently reduce the risk of cross-contamination (see below). Insufficient cleaning can quickly lead to contamination problems for the next product manufactured on the same line.
New Guidelines for Risk Assessments
In line with the new EU GMP Guidelines, Chapter 3 and Chapter 5, a risk assessment has to be performed with regard to potential cross-contamination of one product with another—the objective being assuring patient safety.
The risk assessment should summarize the structural and operational controls that are in place as elements of the facility risk management program to reinforce approved processes to minimize and mitigate the risk to product quality.
Reasons for cross-contamination can be manifold and caused by technical as well as organizational deficiencies. Insufficient cleaning of equipment, poor facility design or inappropriate design of the HVAC system may be reasons, as well as contamination via personnel or primary packing material. But also the design of the production process itself can cause cross-contamination, for example, due to open product handling during transfer or sampling operations in shared plants. It is extremely important to control cross-contamination to levels below the acceptance criteria.
A comprehensive risk assessment process includes the assessment of risks related to environment, health and safety (EH&S risk assessment) and risks related to potential cross-contamination and product quality (GMP risk assessment).
Cleaning validation is one crucial element in risk management to ensure that patient safety is not at risk. The latest guidance from the EMA on health-based limits for cleaning validation is provided in the Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities (3).
It is important to consider the whole material flow, e.g., from dispensing to packaging, personnel flow (gowning, movements from one area to another or to corridors, etc.) and general HVAC and facility layout.
Risk assessment methodologies, such as ICH Q9, should be applied.
One example for a risk assessment methodology is FMEA. This methodology requires that for each process step, the following questions be addressed:
- Probability: What is the probability of a cross-contamination at levels higher than the acceptable health-based limits?
- Severity: What would be the effect of this carryover in the patient, based on the toxicological characteristics of the contaminant and the maximum possible carryover?
- Detectability: How easily would a cross-contamination be detected?
Before the facility is commissioned, a riboflavin test can be carried out to determine how the active substance spreads within an aseptic isolator during the filling process. Filling 1 g riboflavin suspended in 1,000 ml of water into prepared vials in the isolator makes it possible to monitor the spread of the substance during the manufacturing process and subsequent cleaning process, as well as the risk of cross-contamination (4). The test results can then be used to define sampling points for swab tests in the cleaning validation process.
Certain potentially critical situations or operations should also be assessed, and if necessary, simulated:
- Vial breakage in the filling area and during freeze drying
- Vial breakage in critical areas within the isolator, such as vial separation or crimping
- Removal of liquid/powder and vial, and subsequent cleaning in the event of vial breakage
- Unloading of contaminated cleaning equipment
- Connection and disconnection of the HPAPI buffer container in the isolator
Depending on the surface properties of nonproduct contact parts such as plastics used to transfer or separate the vials, it may be necessary to dedicate them to a specific product. The surfaces of plastics are not qualifiable, cleaning cannot be validated, or the effort involved in cleaning is disproportionate in comparison with the use of product-dedicated parts.
Bringing Worker Protection and GMP Together
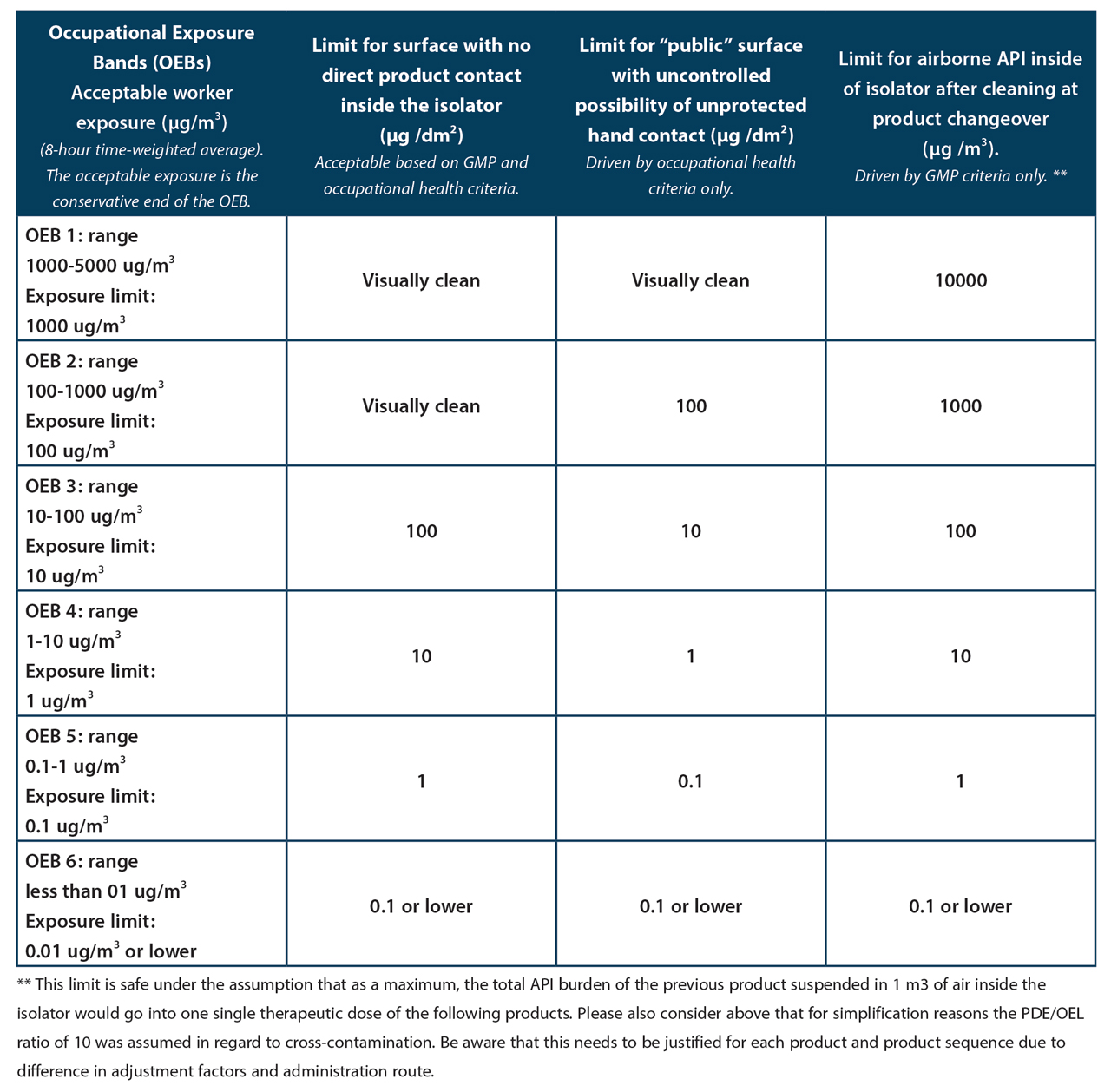
Cleanliness of non-product contact surfaces has always been a topic of relevance for worker protection. The most common route of worker exposure is via hand contact. The surface of the palm of a hand is approximately 100 cm2 (= 1 dm2). This is why surface contaminations are frequently expressed in µg/dm2. The allowable surface contamination is obviously inversely related to the toxicity of the substance, expressed for example as PDE. However other parameters play a role in determining the level of allowable surface contamination such as the assumed frequency of the contact and whether the persons who touch the surface are aware of the fact that it carries residual contamination and are likely to avoid contact as much as possible. Therefore, limit values for “public” surfaces, e.g. handrails in public staircases, office desks, etc. must be more stringent than those where access is limited: walls and floors of production rooms, interior of isolators, etc. To correlate surface contamination with risk to the worker, various models have been developed. A popular one assumes 10 hand contacts per shift and 100% surface-to-skin transfer as well as 100% dermal absorption. This means that based on this model, each dm2 can be contaminated with a maximum of 1/10 of the PDE of the substance in question. This is an extremely conservative model, but this level of cleanliness is often achievable by very good cleaning and can then serve as a rationale to state that cleaning has been successful. In practice, the value derived with this model should only be directly applied as a limit for surfaces that require very high levels of cleanliness.
It is important to note that the default assumptions of this model should be replaced by substance and situation-specific data whenever such data are available.
At the latest with the publication of the revised Chapter 5 of the EMA GMP guidelines on August 13, 2014, GMP has now also become interested in the cleanliness of non-product contact surfaces. The cleaner they are the lower the risk of cross-contamination via the airborne route and via mechanical transfer. The logic of this statement is obvious. It is of greater practical relevance in non-sterile manufacturing where systems are more open than in aseptic or sterile production where systems are generally much more closed for reasons of product protection. Compared to the correlation between surface contamination and risk to the worker, it is even more cumbersome to establish a relationship between the contamination of a non-product contact surface and a cross-contamination risk. It is therefore very difficult if not impossible to establish science-based non-product contact surface limits in the GMP context. Scenarios are sometimes applied where all the substance residues on floors, walls, on the outside of equipment, etc. are assumed to unite and make a targeted and synchronised move into the next batch of a product or even a single dose of the next product. Similar scenarios are also sometimes used to assess cleanliness levels of those parts of the insides of isolators that have no direct product contact.
For worker health protection, the inside of an isolator is not a public surface and workers are aware that such an isolator may carry traces of products handled earlier. Therefore, it is rare to demand work-health driven cleanliness levels lower than 1 PDE per dm2 of surface.
It is proposed that this level of cleanliness should, in the majority of cases and after due consideration of exact circumstances of the individual situation, also suffice for GMP. It means that the whole contamination on 1 dm2 of inside surface of the isolator would have to be transferred into one single daily dose of the next product to cause a patient risk issue. We would like to suggest that this scenario is so unlikely that it can be safely assumed that it will never be exceeded in reality. Where one PDE per dm2 would represent a level of contamination that would still be visible (PDE of several 100 µg /day), the minimum requirement would of course be the one of visual cleanliness (cf. Table 1. Proposed EH&S and GMP surface limits for non-product contact surfaces and air limits inside of isolators).
Summary
Isolators are being used more and more frequently in the sterile manufacture of pharmaceuticals to protect the product from direct access by personnel in the critical manufacturing process. The use of isolators also makes it possible to reduce the size of the GMP-critical area for aseptic manufacture in clean-room Class A / ISO Class 5 to an absolute minimum. Isolators are considered in general to be non-product-contact surfaces as, for the large part, their surfaces and fixtures do not come into direct contact with the product. However, product contact contamination at critical locations within an isolator can occur – in the filling area for example. In theory, contamination of the air due to released product particles can also create a risk. While this applies to all substances manufactured in an isolator, special attention should be paid to HPAPIs, for which the permitted daily exposures (PDEs) are particularly low. The EMA Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities dated November 20, 2014 applies when it comes to setting these PDEs.
References
- Vanhecke, P., Sigwarth, V., and Moir, C. “A potent and safe H2O2 fumigation approach.” PDA Journal of Pharmaceutical Science and Technology 66 (2012): 354–370.
- Denk, R., et al. “Joint Assessment of Occupational Safety and GMP Aspects.” Pharmind 4 and 5 (2017) 492–498.
- Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities (20 November 2014) EMA/CHMP/CVMP/SWP/169430/2012
- “Riboflavin test for low-germ or sterile process technologies.” Information sheet. Mechanical Engineering Industry Association. (December 2007).
About the Authors
-

Richard Denk, SKAN AG
Furthermore, Denk is a member of the PDA Advisory Board for ATMPs, and he is a member of the ISO TC 198 WG-9 Aseptic Isolator Group. Denk founded 15 years ago the Containment Expert Group of the ISPE D / A / CH. The Containment Expert Group published the Containment Manual that Denk was responsible for in September 2021. Denk has spent more than 30 years with the subject production of aseptic processing and highly active/highly hazardous substances and has developed the containment pyramid.
-

Andreas Flueckiger, Roche Group
Andreas is in charge of the Roche Group’s occupational health and hazard assessment programs.
-

Hirokazu Kisaka, Takeda Pharmaceutical Company
Hirokazu joined Takeda Pharmaceutical Company in 1988 and currently works for Takeda as Head of Regional Engineering Japan and Asia.
-

Reinhold Maeck, Boehringer Ingelheim
Reinhold works at Boehringer Ingelheim as Head of Regulatory Intelligence EHS&S. He has more than 20 years of experience in the pharmaceutical industry.
-

Lars Restetzki, Roche Group
Lars is Business Process Manager in Roche’s Rocephin manufacturing plant.
-

Andreas Schreiner, Novartis
Andreas is Head of Validation for Solid Dosage Forms in Novartis’ Manufacturing Science and Technology Organization.
-
Rico Schulze, Landesdirektion Sachsen
Rico is a GMDP Inspector with the local GxP inspectorate in Dresden, Germany. He is also the head of the German authorities’ expert group on radiopharmaceuticals.