

Knowledge-Driven Impurity Control for NMF Protein Products

Product- and process-related impurity clearance and control are central to biopharmaceutical chemistry, manufacturing and controls strategy (CMC) to ensure product safety and efficacy.
To address unmet medical needs, an increasing number of new molecular format (NMF) protein products are entering biopharmaceutical pipelines. These products are characterized by their artificial molecular design, which leads to unusual physicochemical properties. Additionally, they require product-specific manufacturing processes and formulations.
As a result, impurity clearance and control present enormous challenges to the biotech industry. These challenges are significantly greater compared to those associated with classic monoclonal antibody (mAb) products, which benefit from a manufacturing-friendly platform process, standardized formulation and established analytics.
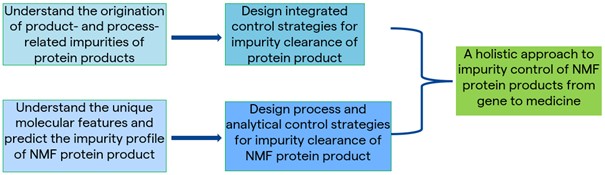
Application of Biopharmaceutical CMC to Design-Integrated Impurity-Control Strategies
ICH Quality Guideline Q6B: Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products clearly defines the specification needs of the composition of protein drug products, the desired products, part of products, product-related drug substance, product-related impurities, process-related impurities and contaminants. As illustrated in Figure 1, besides the misassembly or misfolding inside expression cells due to the molecular design, the product-related impurities usually originate from the interactions of active pharmaceutical ingredient (API) protein molecules with the contacting matrices under the influence of environmental stresses from production in cells and secretion to cell culture media, to downstream purification, formulation, fill and finish, storage, transportation and, finally, to administration to the patient.
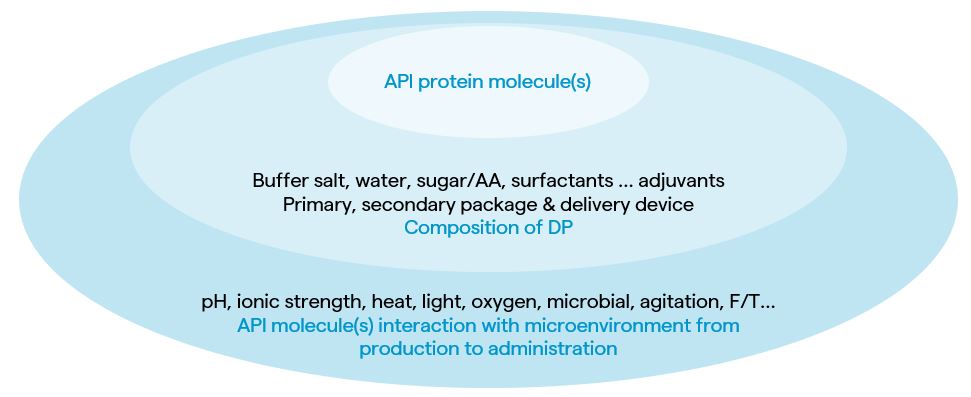
Legend: AA: amino acids, DP: drug product, F/T: freeze/thaw)
As summarized in Table 1, the major product-related impurities and the strategies of prospective knowledge-driven initial process design and analytical development should focus on the potential critical quality attributes (CQA).
| Isoforms/PTMs/Degradation | Liable Sequence/Amino Acids | Impact from Process | Analytical Methods |
|---|---|---|---|
| Chain mis-assembly | KiH other point mutations | Molecular scaffold/format | Intact MS, RPC, HIC |
| Sequence variants | Site mutation, misincorporation, S→N, V→I, | Cell line stability, culture media | Peptide mapping – MS |
| Glycan isoforms | N-X-S/T (X is not P), S, T, Y | Host cell, culture conditions | U/HPLC-MS, sialic acid |
| Aggregates | Surface hydrophobic patch | Buffer/matrix, stresses | SEC, AUC, nrCE-SDS |
| Fragments | N-P/G/S, proteases target site | Buffer, HCP proteases, metal ions | SEC, RPC, rCE-SDS, MS |
| Oxidation | C, M, H, Y, W | Oxidants, free radials, lights, metal ions, temperature | PMAP-MS, icIEF, CZE, IEC |
| Deamidation | N, Q, NG, | pH, bufferm temperature, PO43-, Tris | PMAP-MS, icIEF, CZE, IEC |
| Free Cys | C,S-S scrambling | pH, redox potential | PMAP-MS |
| Interaction with matrix | K, C, H, D, E | Citrate, reducing sugar, Metal ions, temperature, resins, filters | Intact/subunit/PMAP-MS, RPC |
| Other PTMs | Phosorylation on S, T, Y | Intact, subunit, PMAP-MS | |
| Other degradations | Racernization H, D, S Crosslinking K, S, D, E, Q, C | Buffer pH, temperature | Intact, subunit, PMAP-MS |
Figure 2 shows the process-related impurities of protein drug products originating from manufacturing and administration. The recombinant protein-product CMC applies science-and risk-based integrated control strategies for impurity clearance and control. Impurities could be defined as CQAs, based on their impact on safety (including immunogenicity), activity and pharmacokinetics or pharmacodynamics rofiles. The clinically relevant patient-centric specifications for impurity control are set up with a focus on patient safety considering the identity of the impurity, the assay for impurity control, the safety profile of impurity, the process capability of impurity control and removal, and the impact of impurity on product quality. Usually, the integrated impurity-control strategies include the following elements:
- Control of manufacturing environment/facility/equipment
- Control of raw materials
- Control of process parameters
- Leachables/extractables assessment/control
- In-process controls/testing
- Batch-release testing
- Stability testing
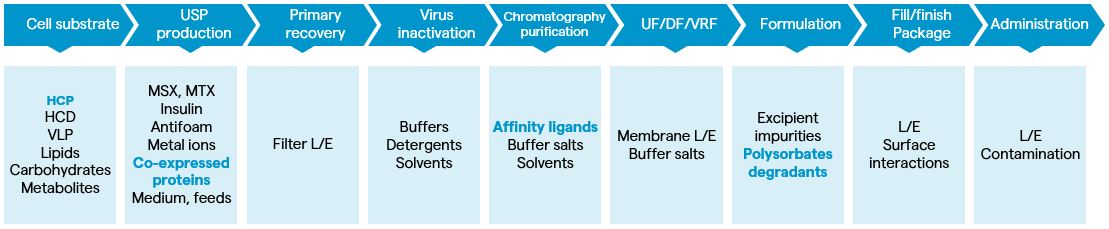
Legend–HCD: host-cell DNA, VLP: virus-like particles, USP: upstream process, MSX: methionine sulfoxamine; MTX: methotrexate; UF: ultrafiltration; DF: diafiltration; VRF: virus- reduction filtration; L/E: leachables and extractables. Impurities originated from raw materials and adventitiously introduced impurities are not shown)
Knowledge-Driven Design of Process and Analytical Control Strategies
In contrast to mAbs consisting of immunoglobin domains evolved from natural selection with desirable stability and solubility for manufacturing, promising NMF protein molecules usually originate from artificial design. They have unique biological functions, with challenging physicochemical properties to manufacture and, consequently, unusual process- and product-related impurities to control.
Due to the restriction from a required amino-acid sequence and high order structure for high specificity and appropriate affinity on biological targets (such as enzyme, receptor, antigens) or on the limitation of resource and time in the early discovery and development stages for sequence optimization and lead-molecule selection, many NMF molecules have unusual physicochemical properties such as high hydrophobicity, low conformational stability, low colloidal stability and low solubility, and they are more prone to aggregation. The impact of low solubility and aggregation on biomanufacturing is well known, but the deteriorating effect of low conformation stability on chromatographic purification is unintentionally ignored because of its poor detectability until a loss of yield or activity are noticed.
Some NMF proteins, such as molecules mainly consisting of single-chain variable fragments or multiple-binding domains, have very high hydrophobicity; consequently, they are liable to nonspecific binding on various contact surfaces. The strong hydrophobic interactions can lead to conformation changes, denaturation of the protein and irreversible product loss. High hydrophobicity increases the chance of a hijack of host-cell protein (HCP), especially lipases with exposed hydrophobic domains to bind lipid substrate, on API protein through purification. Subsequently, this leads to hydrolysis of polysorbate excipients in the liquid formulation, degradation of the product protein and formation of visible and/or subvisible lipid particles in the final dosage form.
Many NMF proteins have a complex multimer structure requiring multiple-gene expression vectors for production with the potential to generate chain misassembly impurities, such as asymmetric 4-chain IgG-like bispecific antibodies.
Some NMF proteins have low-expression titers with high HCP interferences. In worst-case scenarios, the product titer is less than 10% of the HCP (<0.1mg/mL), which puts a heavy burden on the impurity clearance during downstream purification, such as overloading of expensive affinity-capture resins with HCP impurities, resulting in short resin lifetimes and a high cost of goods.
Artificially designed NMF-protein molecules are prone to having non-ideal linking sequences, liable to fragmentation and aggregation.
NMF products originated from endogenous proteins and usually have very complex glycosylation profiles. Major efforts are required for the selection and development of an appropriate expression cell line, development and optimization of upstream process (USP) for obtaining a high-quality product with an ideal glycan profile.
Some NMF products are highly phosphorylated for special biological functions. The negatively charged phosphate groups form strong complexation with exposed metal ions on various surfaces, such as vessels, containers, resin particles, and filter membranes, during the manufacturing process. This leads to product loss, cross-contamination and deterioration of the resin and membrane. An appropriate buffer with competitive metal ion-complexing components is needed to mitigate the problem for efficient manufacturing, such as to confirm cleanability in multiproduct GMP manufacturing facilities. More importantly, the strong complexation of phosphate groups, with metal ions on exposed surfaces, also causes difficulties in analytical methods development because of the strong interaction of protein analytes with the resin/gel matrix and contacting surfaces.
Another unusual feature of NMF-protein products is residues of enriched complexing amino acids in the primary sequence or clustered on the molecular surface. These amino acids form additive complexation with exposed metal ions on various surfaces during manufacturing. Thus, the undesirable complexation causes a range of problems, such as product loss, the need for cleanability assessment, deterioration of chromatography and filtration, and assay failures.
Based on our experiences with many NMF-protein projects, to meet the challenges of CMC development, a knowledge-driven approach to design-integrated control strategies should be initiated from a mechanistic understanding of the product and process, such as the:
- In-depth understanding of the molecule, structural liabilities and physicochemical properties
- Predicted product-related impurity profile
- Predicted process-related impurities
- Interaction of product protein with process matrices
- Intended medical use of the product (e.g., the required purity of products for ocular administration is much higher than general intravascular administration)
A Science- and Risk-Based Holistic Approach for Proactive Control of Product- and Process-Related Impurities
A holistic approach for proactive impurity clearance should start with the design of the molecule. Ideally, the design will result in a molecule that is easy to manufacture, with minimal impurities and maintains desirable biological function. If possible, attempts should be made to maximize the physicochemical differences between the desired product and the potential impurities for feasible purification and monitoring.
Optimization of lead selection via an adequate developability exercise to avoid physicochemical-instable molecules should be a routine practice for the development of complex NMF-protein products.
Optimizing vector design, cell-line construction and USP to improve product quality, increase titer and decrease downstream-related impurities (DSP) will make purification, formulation and analytical control much easier. This is in line with the quality-by-design strategy in ICH quality guidelines. The CMC development of NMF-protein products should start with the end in mind. As long as it is practically possible, the design of a molecule and product and the effective manufacturing process with minimal product- and process-related impurities will build the solid foundation for impurity clearance and control.
Rational design and systematic optimization of DSP are critical for the effective clearance of product-related impurities and USP-related impurities as well as minimizing DSP-related impurities. Specifically, the clever selection of chromatographic resin, filter, membrane and buffer conditions that are compatible to relatively unstable NMF-product proteins is critical in impurity control.
The rational design of formulation for product stability is key to preventing the generation of new impurities during storage, transportation and administration. The formulation also should be suitable for drug product manufacturing, dosing regime and administration defined by a quality target product profile (QTPP), such as high concentration and low viscosity for subcutaneous injection.
An appropriate strategy for drug product manufacturing, from compounding to fill-finish and packaging, is essential for impurity control of the final dosage form of NMF-protein products.
The design of a fit-for-purpose analytical control strategy should be based on the prediction of potential process- and product- related impurities, with due diligence to the intrinsic properties of the protein-molecule and its interactions with microenvironmental matrices. For NMF-protein products, early and rapid development and validation of product and process specific- analytical methods are usually based on the predicted impurities that are potential CQAs of the product.
Conclusions
NMF proteins have unique structural features and challenging physicochemical properties. A holistic approach is required to design and develop effective and efficient process and analytical control strategies for product- and process-related impurity clearance and control. The rational design of integrated impurity control strategies should be proactively knowledge-driven and based on a mechanistic process and product understanding:
- In-depth understanding of the molecular-structure liabilities and physicochemical properties
- Predicted potential product-related impurity profile
- Product-specific manufacturing process from gene to medicine
- Intended medical use of the product
About the Author
-

Qifeng Zhang, PhD, Lonza, Lonza Biologics
Zhang graduated with a Bachelor of Science in biology (1993) and a PhD in analytical organic chemistry in 1998. Zhang is currently a Global Senior Technical Lead at Lonza, focusing on understanding the molecule to design knowledge-driven CMC strategies for products, processes, and analytical development of novel molecular format protein products from lead molecule optimization to commercial release.