

Manufacturing Capacity Expansion and Validation for Autologous Cell Therapies

Autologous chimeric antigen receptor (CAR-T) cell therapies have the potential to revolutionize the approach to treating hematologic malignancies.
These therapies modify the patient’s own T-cells to recognize and destroy cancer cells. While these novel therapies are changing the landscape for successful cancer treatment, there are significant challenges that exist in the manufacturing scale-up and validation compared to traditional biologics.
Capacity expansion for autologous cell therapies is complex due to single-patient manufacturing. Unlike traditional biologics, where one drug product batch can typically dose hundreds or thousands of patients, autologous cell therapy batches are manufactured for single-patient use. Dosing significantly more patients in late-stage clinical trials and commercial manufacturing can typically only be achieved by well-planned, proportional expansions of manpower, manufacturing and testing facilities, and all their supporting functions. These unique requirements can create roadblocks to increasing capacity output rapidly and reliably to supply cell therapies to more patients (see Figure 1).
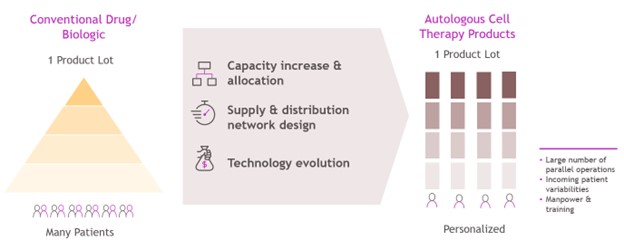 Figure 1 Manufacturing and supply-chain differences between CAR-T products and conventional drugs and biologics
Figure 1 Manufacturing and supply-chain differences between CAR-T products and conventional drugs and biologics
 Figure 2 Operational challenges unique to the supply-and-demand balance for autologous CAR-T products
Figure 2 Operational challenges unique to the supply-and-demand balance for autologous CAR-T products
Additional operational complexities for autologous cell therapy products exist that further require long-term planning and investment decisions for manufacturing output capacity expansion(s). Raw material supply shortages (viral vector), demand variability, patient cancellations (also a potential delay or replacement), out-of-specification drug products and patients requiring re-apheresis (delayed planned manufacturing) can impact a cell-therapy supply-chain supply-and-demand balance (see Figure 2). Expanding capacity in a way that allows the maximum number of patients to be served while also preventing excess capacity carrying costs is crucial.
Also unique to cell therapy are potential penalties for not delivering the established capacity, both by regulatory agencies and patient communities. Drug product material must be delivered on time and according to specifications, and failing to deliver on the capacity commitments can result in a loss of reputation, trust and revenue.
Various options exist for manufacturing-capacity expansion, and each requires different levels of validation depending on the nature and extent of the change. Capacity validation is the process of demonstrating that any changes or additions to existing manufacturing do not lead to higher manufacturing deviations and/or product quality risks. Prospective capacity-validation studies ensure that manufacturing capability, including turnaround times and product quality, remains non-inferior when compared to pre-expansion data.
Options for Expanding Manufacturing Network and Validation Required
Short-term and long-term options exist for capacity expansion that vary in capital cost, time, output and validation studies required. When expanding a manufacturing network of CAR-T therapies, there are five common methods for capacity expansion (see Figure 3).
Short-term options are ideal when quick, cost-effective capacity expansion is needed. However, the increase in throughput volume is often limited. These expansion options include increasing the capacity of existing suites or rooms and adding suites or rooms to an existing site. Since these increases are made within preapproved sites or suites, the capacity validation studies and conditions are typically less rigorous.
On the other hand, long-term options are beneficial when a substantial increase in throughput volume is expected. These options often have longer lead times for implementation and require more extensive capacity-validation studies and acceptance conditions. Long-term options include expanding an existing site, adding an internal site or using a contract manufacturing organization (CMO). These may require construction, technology transfer and extensive validation and/or comparability studies, especially since they involve new sites and facilities that have not yet been approved. Different validation methods and data requirements are needed as well, depending on the nature of the capacity expansion.
 Figure 3 Operational challenges unique to the supply-and-demand balance for autologous CAR-T products
Figure 3 Operational challenges unique to the supply-and-demand balance for autologous CAR-T products
Increase Existing Suite/Room Capacity
There are multiple options for increasing the capacity in a room or suite that is already approved by a regulatory agency. A few options are:
- Optimizing the layout of the manufacturing space: Efficient use of the available space can allow for additional equipment to be added.
- Decreasing turnaround time: Streamlining processes to reduce the manufacturing process time.
- Automating processes: Automation can increase efficiency, reliability and speed.
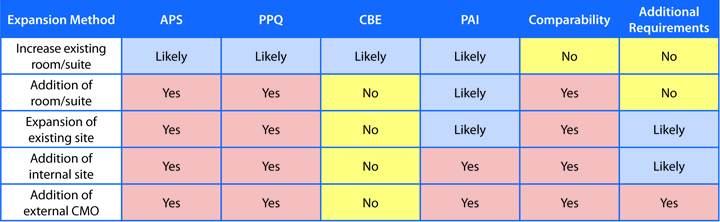
Depending on the change, aseptic process simulation (APS), process performance qualification (PPQ), change being affected (CBE) and/or pre-approval inspection (PAI) may be required. It is unlikely that comparability or other requirements will be needed. However, they could still provide valuable information for process understanding and control. Therefore, their inclusion should be considered using a risk-based approach.
Addition of Suites/Rooms to an Existing Site
Adding suites or rooms to an existing approved site requires several validation activities. The new room must be proven to be sterile, which is typically achieved through the re-execution or modification of the APS. Depending on the significance of the room addition, the regulatory agency may require a PPQ. A CBE filing is required if it aligns with the conditions of the previous agreement from agency interaction. If the change is within the Post-Approval Change Management Protocol (PACMP) framework, a CBE filing is typically appropriate. However, if the change is outside of the PACMP framework, a Prior Approval Supplement (PAS) is typically expected. These activities ensure that adding the new suites to an approved site complies with regulatory requirements and maintains the established manufacturing conditions and product quality and safety of the cell-therapy product.
Expansion of Existing Sites
The expansion of existing manufacturing sites for cell therapy is a complex process that can involve either the addition or expansion of rooms in an already approved manufacturing building, or the construction of an entirely new manufacturing building. Given the significant nature of these changes, comprehensive validation requirements are often necessary. These include APS, PPQ and comparability studies. In addition, a PAS and/or PAI may be required. It is crucial that the expanded site demonstrates that the manufacturing process and output are consistent with already-established, reliable site conditions, ensuring the quality, safety and supply of the cell-therapy products.
Addition of Internal Site
The addition of an internal site, one that is not already in operation or does not have regulatory approval, is a long-term strategy for expanding capacity in cell therapy. This can be achieved through a merger, acquisition or construction. Opting for an internal site over an external site offers more control over operations and future expansions. The validation requirements for such an expansion are comprehensive and include APS, PPQ, comparability studies and a PAS.
Addition of External CMO
Incorporating an external site that does not currently have regulatory approval for the product to be manufactured is a long-term strategy for increasing capacity. This approach can potentially reduce initial capital investment and expedite time to market, especially if construction is not required. However, the organization may have less control over operations, and contracts, quality agreements and further expansion conditions can be inflexible for the sponsor. Comprehensive validation requirements, including APS, PPQ, comparability studies and PAS are required. Other requirements are also likely to be needed, depending on the specific nature of the site.
The authors have provided major highlights of the challenges autologous cell-therapy supply-chain organizations face when selecting options for capacity expansion and validation throughout development and in commercial operations. There are multiple options for increasing capacity with different expectations for capacity validation studies, implementation times (including approval), benefits (capacity increase), costs and validation requirements to allow additional patients to safely receive treatment. Current capacity-validation studies are focused on manufacturing, and sponsors should also carefully consider the proportional increased demand for drug-product-testing. The authors hope that this article will stimulate more industry guidance in the future to support efficient cost-benefit capacity expansions to maximize patient access to life-saving cell-therapy treatments.
About the Authors
-

Stephan Krause, PhD, Bristol Myers Squibb
Krause, PhD, Executive Director for Cell Therapies at Bristol Myers Squibb and Chair of PDA’s ATMP Advisory Board, is a results-driven leader with technical, managerial, and executive experience. He has a proven background in leading quality and technical functions within global operations. Krause is a recognized leader of industry working groups to improve and standardize best practices for analytical and quality-focused processes. His breadth of experience includes mAbs, ADCs, ATMPs, vaccines, coagulation factors, small molecules and biosimilars
-

Adam Boyer, Bristol Myers Squibb, Bristol Myers Squibb
Boyer, CSCP, is a dedicated Cell Therapy Supply Chain Manager at Bristol Myers Squibb. He is committed to continuous learning and is eager to contribute to the advancement of cell therapy through strategic supply chain management.