

Validating NGS-Based Assays In-House for Virus Safety Assessment A Call for Action

The fruitful discussion that transpired from Izabela Fabianska’s and my presentation at the PDA Next Generation Sequencing Workshop 2024, “The Robustness of NGS Analysis Pipelines for Virus Detection in Biologics,” and our participation in the insightful PDA Virus Conference 2024, left us very excited. It inspired me to summarize my key takeaways from my time in Amsterdam and share my conclusions on the requirements for operating a good manufacturing practice (GMP) platform for next-generation sequencing-based assays. To extend the conversation and delve deeper into these topics, we have launched a podcast series that brings together voices from the industry to foster an exchange of ideas within the field (1).
Validating NGS-Based Assays in House: The time is now!
Next-generation sequencing (NGS) is a highly sensitive method for evaluating biotherapeutics' quality and virus safety, from monoclonal antibodies and cell-and-gene therapeutics to vaccines that have become increasingly accepted in the biopharmaceutical industry. Guidelines are being updated by regulatory bodies for biopharmaceutical companies on the application of NGS-based assays within a GMP environment. Competent authorities, like the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA), have implemented the International Council for Harmonisation (ICH) Quality Guideline Q5A(R2): Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin (2-4). Furthermore, the European Directorate for the Quality of Medicine & Healthcare is drafting a new European Pharmacopoeia (Ph. Eur.) 2.6.41 chapter centered on high-throughput sequencing for the detection of viral extraneous agents (5).
Validating NGS-based assays is a significant step forward in aligning with the evolving regulatory landscape. By validating these assays in-house, companies can retain control over their processes, ensuring that the stringent requirements for accuracy, reliability and consistency are met. This provides more flexibility and adaptability in response to changing market needs and technologies. Additionally, the validation of NGS-based assays internally—as opposed to outsourcing—can lead to more streamlined and secured workflows, as it eliminates the reliance on external entities for GMP-compliant data analysis and results delivery. This can lead to cost reductions, assays tailored to internal processes and protocols, faster turnaround times and better protection of intellectual property (see Table 1). Driven by these benefits, the trend toward internal validation of NGS-based assays signifies a new benchmark in biopharmaceuticals with the potential to improve the safety and effectiveness of biotherapies. As we continue to advance in this direction, we can expect to see even greater improvements in patient care and therapeutic outcomes.
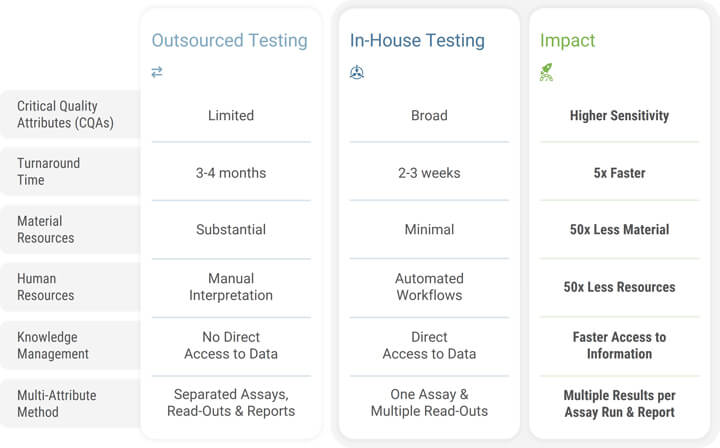
The Established Implementation of GMP Processes in Biopharmaceutical Organizations
Assay validation, a critical component of GMP in the biopharmaceutical industry, is a systematic process that showcases the accuracy, reliability and consistency of an analytical method. This regulatory framework is used to evaluate the quality and safety of biotherapies. The validation process is critical to ensure that the analytical methods used in GMP environments yield trustworthy results. This process involves several steps: confirming selectivity, evaluating accuracy, specificity, linearity and precision, and determining a valid range for conducting the assay (2). Biopharmaceutical companies are committed to maintaining the integrity and traceability of their products, ensuring that every batch of a drug is safe and of the highest quality. This commitment is in line with fulfilling GMP regulations, which play a significant role in assuring drug quality and patient safety.
Scaling Up the Potential of NGS-Based Assays: A Work in Progress
Next-generation sequencing has developed into an indispensable tool for ensuring the quality and safety of biotherapies. It offers a rapid, highly sensitive and unbiased approach to DNA and RNA sequencing, delivering cost-effectiveness in terms of per-base sequencing expenses. NGS allows for the identification of potential viral contaminations, variants and novel viruses, thereby ensuring the safety and integrity of biotherapeutics. NGS is anticipated to replace traditional assays, such as in-vivo animal tests and supplements, and/or replace in-vitro virus safety tests, enhancing the safety of the entire manufacturing process. NGS has the potential to significantly contribute to the viral safety evaluation of biotherapies, a critical aspect in producing safe and effective treatments such as monoclonal antibodies, cell-and-gene therapy drug products and vaccines. Its high sensitivity and broad detection potential make it an excellent tool for testing cell banks (including the master and working cell banks, which are cells at the limit of in-vitro cell age), virus seeds or harvests (unprocessed bulk, drug substance, or product).
In the context of its application in a GMP setting, the publication of the European Federation of Pharmaceutical Industries and Associations (EFPIA), Considerations for Validation and Implementation of Next Generation Sequencing for Adventitious Virus Detection for Biological Medicinal Products, provides valuable insights. It includes a great summary of the technology itself, highlights an approach for method validation, including bioinformatic analysis (the replacement of existing methods) and summarizes the regulatory landscape thoroughly (6). In short, NGS is crucial to ensuring patient safety and therapeutic efficacy and delivering drugs to patients in a shorter time.
The Remarkable Rise of Regulatory Acceptance in NGS-Based Testing
The potential of NGS in the pharmaceutical industry is increasingly evident, as exhibited by the implementation of regulations and guidelines for its use in GMP, which is currently underway. The benefits of this technology can be fully harnessed once the validation is complete. In 2018, the FDA issued two guidelines for the design, development and validation of NGS-based testing (5). The first guideline emphasizes the use of FDA-recognized databases to reinforce the clinical validity of NGS-based tests (8), while the other provides key insights into the design and development of NGS-based tests, simplifying the premarket review process and aiding in diagnosing suspected germline diseases (9). These guidelines provide a framework for the agency to streamline the development and review of NGS tests.
The ICH Q5A(R2) guideline, adopted by both the FDA and EMA in 2024, describes the testing and evaluation process to ensure the viral safety of biological products and specifies the required data for product registration. The guideline encourages the use of NGS technologies to replace in-vivo assays due to its proven breadth of detection and sensitivity, and the known limits of in-vivo tests. Q5A(R2) suggests that NGS can be used to supplement and/or replace in-vitro cell culture infectivity assays. This recommendation contributes to the reduction of animal use and timelines (10-11). However, before the implementation of NGS in a GMP environment can be considered, a comprehensive validation package of the NGS workflow must be presented to the authorities. This package should comprise the entire NGS workflow—from wet lab protocol to lab procedures and data analysis—and prove both the breadth of detection and the sensitivity (i.e., detection limit) of the NGS method as a limit test (6,12).
The new Ph. Eur. 2.6.41 chapter currently being drafted aims to provide a detailed guide for validating NGS for detecting adventitious viruses. It will cover the whole process, including sample pretreatment, nucleic acid extraction, enrichment, library preparation, sequencing, bioinformatic analysis, reporting and follow-up investigations. It will also highlight the process of how to validate an NGS-based assay for the detection of adventitious viruses. The limit of detection and the specificity (breadth of detected viruses) must be determined using standards provided by the regulatory authorities (5). When stepping into a GMP-regulated setting, organizations encounter significant challenges. In the pursuit of validating a diverse range of NGS assays, biopharmaceutical companies are not only challenged by the variability in methods, instruments and protocols but also by the need for robust computerized systems. These systems, through computerized system validation (CSV), ensure the consistent and reliable performance of the assays, thereby bridging the gap between innovation and stringent validation standards.
Computerized System Validation and NGS-Based Assay Validation Are Streamlined Processes
CSV is one crucial component of the general assay validation procedure that pharmaceutical firms must undertake to meet the quality assurance and regulatory requirements and comply with GMP operations. CSV is used to test, validate and qualify a computerized system, ensuring it consistently and reliably performs its intended functions. As a prerequisite for digitalization, CSV facilitates the transition from traditional paper-based documentation systems to computerized systems. It enables the recording of activity and the creation of traceable and immutable data trails that are securely stored and can be retrieved at any time. Digitalization is an exhaustive, systematic and cross-disciplinary process requiring careful planning, diligent effort and documentation to guarantee that the computerized systems are validated and can be used confidently within the GMP environment.
The Code of Federal Regulations under FDA Part 11, along with the European Union (EU) GMP Annex 11, outlines the requirements for validation, audit trails, legacy systems, record copies and record retention, all aiming to ensure secure and dependable records (13). Another significant guideline, ISPE GAMP® 5: A Risk-Based Approach to Compliant GxP Computerized Systems (Second Edition), offers a structural framework that emphasizes risk-based validation to ensure the reliability, consistency and compliance of computerized systems (14). Notably, it also reflects the principles of the revised EU GMP Annex 11. Key components of the qualification process for computerized systems are the documented and executable installation qualification (IQ), operational qualification (OQ) and performance qualification (PQ) protocols. The IQ verifies the correct installation of the software and hardware according to the specifications. The OQ, also known as functional verification, involves testing system functionalities to confirm they operate correctly as specified. PQ is the validation's final step, focused on demonstrating that the computer system operates as anticipated, suits the intended purpose and fulfills the user’s requirements.
On the other hand, the implementation of NGS-based assays in a validated environment presents its own challenges, and the complexity of handling NGS data is one of them. A scalable solution is required to manage the control process for future sample throughput. Outsourcing these assays under GMP to third parties can be an option, but clients might face challenges such as losing control over the process, inconsistent turnaround times and the need to disclose confidential data. Implementing an in-house NGS platform and a scalable software solution that supports the development of multiple NGS assays and GMP operations can be an ideal solution to overcome these challenges. This approach, despite its initial complexity, offers advantages such as complete automation, simplification of assay development and validation, improved user experience, consistent data analysis and management, and optimization of operations across various modalities, applications and facilities. By adopting this strategy, an organization can save time and money, protect intellectual property and streamline operations. Additionally, establishing NGS assays in-house provides a unified and scalable platform for all NGS instruments. This reduces complexity and increases efficiency by enabling real-time collaboration on the same data across different geographies within the organization.
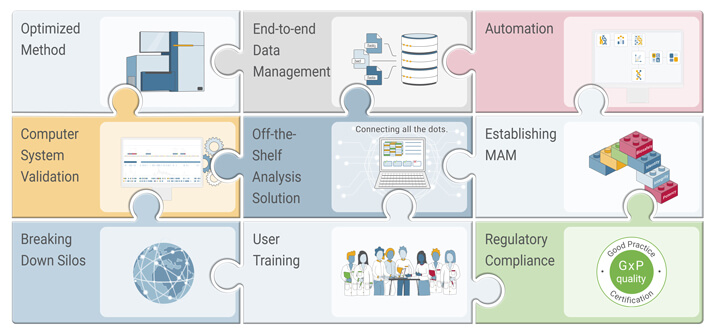
Another trend is to establish NGS assays and software platforms to develop modality-specific critical quality attribute and multi-attribute method (MAM) packages (see Figure 1). These can be swiftly transferred from late-stage development to manufacturing sites across the globe. By leveraging existing knowledge and synergies within an organization, the deployment of local solutions will be streamlined. This approach not only increases quality but also minimizes risk and reduces time-to-patient, especially for gene-and-cell therapeutics (12).
Conclusion and Outlook
NGS is changing the way biopharmaceutical drugs are developed and manufactured. This has led regulatory authorities to establish more detailed guidelines for applying NGS-based assays, including recommendations for validating systems to be used within a GMP environment to ensure the integrity of data assays and reports. By implementing in-house NGS assays in a validated environment, biopharmaceutical companies can improve the sensitivity and specificity of the characterization and release of the drug products while ensuring compliance with quality assurance requirements in a remarkably short time.
Looking ahead, we anticipate that NGS will gradually replace traditional animal testing for virus safety, marking a significant stride in ethical and efficient drug development. Thus, in-house NGS validation emerges as a strategic choice for biopharmaceutical companies aiming to fully harness the potential of NGS in a compliant and efficient manner. The time is now to implement in-house NGS assays, such as adventitious virus detection, and run it in a GMP-compliant environment. This move sets the stage for the future of modality-specific NGS-based MAM assays, transitioning from development to GMP.
References
- Genedata. Spotify Podcast: Industry Voices. https://open.spotify.com/show/24IDewKhWXpygVgznjL8Pc?go=1&sp_cid=c9262c7414b49873f39b59576f53ec61&utm_source=embed_player_p&utm_medium=desktop&nd=1&dlsi=b32b154ac03f4d40.
- International Council for Harmonisation. Quality Guideline Q5A(R2): Viral Safety Evaluation of Biotechnology Products Derived From Cell Lines of Human or Animal Origin. https://database.ich.org/sites/default/files/ICH_Q5A%28R2%29_Guideline_2023_1101.pdf
- U.S. Food and Drug Administration. Guidance for Industry: Q5A(R2) Viral Safety Evaluation of Biotechnology Products Derived From Cell Lines of Human or Animal Origin; 2024. https://www.fda.gov/media/163115/download.
- European Medicines Agency. ICH Q5A(R2) Guideline on Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin; 2024. https://www.ema.europa.eu/en/ich-q5ar2-guideline-viral-safety-evaluation-biotechnology-products-derived-cell-lines-human-or-animal-origin-scientific-guideline
- European Directorate for the Quality of Medicines & Healthcare. High-Throughput Sequencing for the Detection of Viral Extraneous Agents (2.6.41)–Draft. Pharmeuropa (36.2). https://pharmeuropa.edqm.eu/app/phpa/search/.
- European Federation of Pharmaceutical Industries and Associations. Considerations for Validation and Implementation of Next Generation Sequencing for Adventitious Virus Detection for Biological Medicinal Products; 2024. https://www.efpia.eu/media/t22f5yoz/efpia-ngs-virus-detection-paper_finaljun2024.pdf.
- Müllberg, J. Rapid Methods for Adventitious Virus Detection Acceleration of Clinical Timelines/“Speed to Patient”; Am Pharm Rev, 19 Oct 2020.
- U.S. Food and Drug Administration. Guidance for Industry: Process Validation: General Principles and Practices; 2011. https://www.fda.gov/files/drugs/published/Process-Validation--General-Principles-and-Practices.pdf
- Luh, F.; Yen, Y. FDA Guidance for Next Generation Sequencing-Based Testing: Balancing Regulation and Innovation in Precision Medicine. npj Genomic Med 3. 28; 2018. https://www.nature.com/articles/s41525-018-0067-2.
- U.S. Food and Drug Administration. Guidance for Stakeholders and Food and Drug Administration Staff: Use of Public Human Genetic Variant Databases to Support Clinical Validity for Genetic and Genomic-Based In Vitro Diagnostics. https://www.fda.gov/media/99200/download.
- U.S. Food and Drug Administration. Guidance for Stakeholders and Food and Drug Administration Staff: Considerations for Design, Development, and Analytical Validation of Next Generation Sequencing (NGS)-Based In Vitro Diagnostics (IVDs) Intended to Aid in the Diagnosis of Suspected Germline Diseases. https://www.fda.gov/media/99208/download.
- BioPhorum. Recommendations for Establishing a Next-Generation Sequencing Method in a GMP Setting to Confirm the Identity of a Recombinant AAV (RAAV) Gene Therapy Drug Product Recommendations for Establishing a next-Generation Sequencing Method in a GMP Setting to Confirm Identity of a Recombinant AAV (rAAV) Gene Therapy Drug Product; 2024. https://www.biophorum.com/download/recommendations-for-establishing-a-next-generation-sequencing-method-in-a-gmp-setting-to-confirm-the-identity-of-a-recombinant-aav-raav-gene-therapy-drug-product/.
- U.S. Food and Drug Administration. Guidance for Industry: Part 11, Electronic Records; Electronic Signatures – Scope and Application; 2003. https://www.fda.gov/media/75414/download.
- ISPE. GAMP® 5: A Risk-Based Approach to Compliant GxP Computerized Systems (Second Edition); 2022. https://guidance-docs.ispe.org/doi/book/10.1002/9781946964571.
About the Author
-

Christoph Bredack, PhD, Genedata
Christoph works for Genedata Selector in the European biopharma industry, and he focuses on implementing Selector solutions for modality-specific MAM packages during the late stages of analytical development and facilitating their transition into GMP-compliant processes. Christoph represents Genedata Selector within the Advanced Virus Detection Technologies Working Group (AVDTWG) and has contributed to the Swiss ICHQ5A review process. Christoph's European network also contributes to the Industry Voices Podcast series, which started in June 2024. Before joining Genedata, Christoph worked at Roche in Switzerland as a Postdoc in the Neuroscience department of the Molecular Imaging group. Lastly, Christoph earned his PhD in molecular biology and molecular neuroscience at the Max-Planck-Institute of Experimental Medicine in Göttingen, Germany.