

Lifecycle Approach Wipes Away Cleaning Validation Concerns
Cleaning validation is a perpetual undertaking for multiproduct drug manufacturing companies, particularly those with dynamic product profiles and frequently changing commercial needs. With rising demands for complex molecules and highly potent drugs, manufacturers now must continuously invest in new technologies such as containment systems, which offer protection to both operators and finished product (1). This results in manufacturers relying on multiple types of production equipment and manufacturing lines, ranging from production facilities with antiquated technologies (i.e., legacy equipment) to facilities with newer technologies, e.g., isolators or restricted access barrier systems (RABS).
Naturally, using different types of equipment presents a challenge when it comes to successful cleaning validation. One multiproduct manufacturer of generic injectables took a lifecycle approach to cleaning validation, resulting in a cleaning validation program tailored to each specific production line.
Multiple Lines, Multiple Reqs
The manufacturer’s four production lines and their differing requirements for product-based contamination control are summarized in Table 1.
Equipment in all manufacturing facilities is classified as direct, indirect or nonproduct contact. Direct product contact equipment, such as formulation tanks are cleaned using semiautomated procedures, as shown in Lines B and C, whereas, in Lines A and D, formulation tanks are cleaned manually. Similarly, filling equipment is cleaned using automated washers for Lines B and C, and manually for other lines. Isolators are cleaned manually in each line.

As the company expanded, new stateof-the-art production Lines B and C were added, dedicated to noncytotoxic and cytotoxic products respectively. Each line is equipped with a dedicated clean-in-place (CIP) skid and a parts washer for automated cleaning to prevent cross-contamination by actives from other production lines. Lines B and C are also equipped with formulation and filling isolators which are critical to contamination prevention of the finished product.
A sitewide cleaning validation program was developed and implemented in phases, beginning with Lines B and C, followed by Line D. T he program was then extended to dedicated equipment on Line A, the legacy aseptic fill line. The program was designed to follow a three-stage lifecycle approach consisting of 1) Process Design, 2) Process Qualification and 3) Continued Process Verification (2,3).
Stage 1 – Process Design
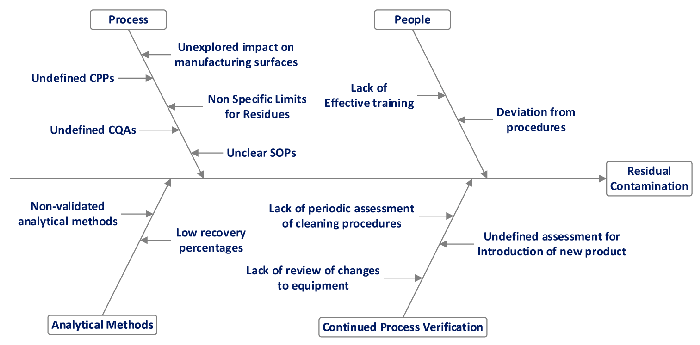
The design space for the cleaning validation program included risk factors associated with products and cleaning procedure variability as this could lead to residual contamination between subsequent batches. A cause-and-effect diagram, highlighting potential causes of failure in the cleaning procedure, is shown in Figure 1.
A cleaning validation masterplan was developed, recognizing the risk factors shown in Figure 1 along with accompanying procedures to adequately mitigate these risks, while defining relevant critical process parameters and critical quality attributes (3,4). The masterplan included establishment of a cleaning validation committee with defined responsibilities for each participating department and cleaning validation program deliverables, leading up to the continuous process verification program stage.
Lines B and C contain portable process equipment cleaned in dedicated cleaning areas using CIP skids for manufacturing tanks and a parts washer for smaller equipment. Operations, such as equipment disassembly and loading parts in the parts washer, are limited to the cleaning area; cleaned and dried equipment is stored in a separate area to avoid any possible cross-contamination between unclean and cleaned parts.
Cleaning cycles developed for CIP skid(s) and parts washer(s) consist of three phases: prewash, cleaning and drying.
The cleaning process for every product is evaluated for effectiveness through visual inspection of product contact areas and testing of rinse and swab samples at the end of the cleaning cycle.
Specific analytical methods such as high performance liquid chromatography or ion chromatography are used to detect residues from production equipment. Swab samples are collected from locations that are either difficult to clean or critical for production. Riboflavin studies determine areas which might be harder to clean than others. Residual limits for swab samples are derived via therapeutic dosages or Acceptable Daily Exposures/Permitted Daily Exposures and calculating Maximum Allowable Carryover (mg) and Surface Area Limit (SAL, µg/cm2) between two batches (5). Recovery factors obtained from method validation studies are also considered for setting product limits.
Additional nonspecific analytical tests such as conductivity and total organic carbon (TOC) are also executed on rinse samples. These provide comprehensive evaluation of tank cleanliness for residues otherwise not analyzed by HPLC/IC, etc. Microbial contamination is assessed through bioburden and endotoxin testing.
The dynamic product profile of the company includes products of varying solubilities, pH levels and toxicities. All products are evaluated for their cleanability by the current cleaning cycle, and, if need be, a newer cycle may be developed to clean a product which cannot be grouped with existing products.
Products filled in Lines B and C are grouped by shared equipment. These products are then ranked based on SAL µg/cm2 between subsequent batches in any possible combination of production schedule.
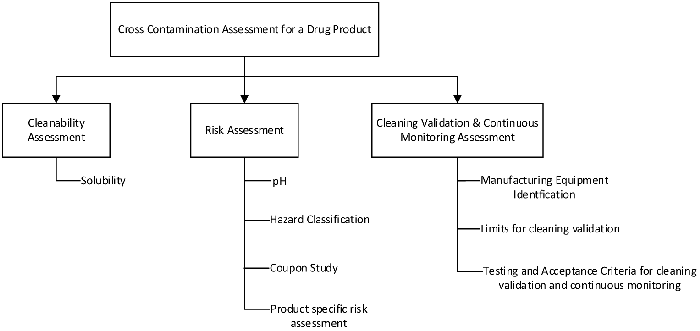
In addition to product ranking, a crosscontamination assessment is done for products, as shown in Figure 2, before introducing a product into an existing matrix. Through this cross-contamination assessment, conclusions are drawn regarding whether equipment may be shared, or whether dedicated or disposable equipment should be used. In addition, cleaning validation approaches for direct contact parts and verification and monitoring requirements for indirect and nonproduct contact surfaces are determined.
Coupon studies are executed as a part of cross-contamination assessment for all new products to evaluate product cleanability as well as potential product impact on the intended manufacturing equipment surface. Typically, coupons are soiled with a product and dried in air for a minimum of 72 hours before proceeding with cleanability assessment. Products with low pH are particularly susceptible to rouging. When this occurs, a requirement to clean equipment immediately postproduction or use disposable equipment is considered.
Stage 2 – Process Qualification
Cleaning validation runs are routinely scheduled to coincide with production runs. If necessary, equipment may also be soiled artificially and dried for a cleaning validation run. At least one cleaning verif ication run is executed on each product, depending on its risk profile and studies executed during the process design stage (6). If warranted, a minimum of three cleaning validation runs are performed on high-risk products.
Validation runs are executed on both shared and dedicated equipment. For shared equipment, limits are calculated for all products manufactured using the equipment and the cleaning process is validated for the lowest limit.
Stage 3 – Continuous Process Verification
Risk factors associated with continuous process verification can be seen in Figure 2. Amultifold approach is implemented to ensure that the cleaning process remains in validated state.
- Automated cleaning processes are verified periodically through a cleaning verification run. Visual inspection and inline rinse monitoring are performed at the end of each cleaning event.
- For manual cleaning processes (i.e., dedicated or nondedicated equipment) at the end of each cleaning event, cleaning is verified through visual inspection and rinse sampling for TOC to ensure there are no anomalies in the cleaning process.
- All changes to equipment or manufacturing process flow are assessed for their potential to impact the cleaning process and these assessments are captured through change controls to ensure traceability. If cleaning validation/ verification is to be executed on new equipment, a worst-case product and manufacturing condition that applies to the equipment is chosen, and the run is executed with rinse sampling for residual and microbial assessment and swab sampling for product specific residual assessment.
Conclusion
Aimed at robust product quality, safety and patient protection, a successful cleaning validation program requires a lifecycle approach. Multiple processes, tailored to suit facility design, technological opportunities available in each production line and variability of the product profiles are required to support an expanding product portfolio in a multiproduct facility. Risk assessments evaluate the cleanability and detectability of the product, leading to procedures that prevent cross-contamination among batches.
[Editor’s Note: The online version has additional content.]
References
- Mirasol, F. “Automation Trend in Fill/Finish Reduces Contamination Risk.” BioPharm International 31 (2018) 12–13.
- Guidance for Industry, Process Validation: General Principles and Practices, January 2011, FDA
- Snee, R.D. “Building Blocks of Quality by Design.” IVT Network (Jan. 24, 2013)
- ICHQ8(R2)
- Technical Report No. 29 (Revised 2012): Points to Consider for Cleaning Validation
- Altmann,T., et al. “Developing A Science-, Risk-, & Statistics-Based Approach To Cleaning Process Development & Validation.” Pharmaceutical Online (June 28, 2017).
About the Author
-

Raji Vathyam
Vathyam has nearly ten years’ experience in aseptic processing in pharmaceutical manufacturing including sterile injectable and medical devices.